06/05/2025
La química orgánica es un vasto universo de moléculas con estructuras intrincadas, y para explorarlo, los científicos necesitan herramientas poderosas que les permitan "ver" cómo están organizados los átomos. Una de las técnicas más revolucionarias y ampliamente utilizadas para este propósito es la Resonancia Magnética Nuclear (RMN), y en particular, la RMN de Protón (1H-RMN). Lejos de ser una simple tabla de números, un espectro de RMN es un mapa detallado que nos cuenta la historia de cada protón en una molécula. Pero, ¿cómo se traduce esa historia en información estructural precisa? La respuesta reside en un concepto fundamental: el desplazamiento químico.
Acompáñanos en este viaje para desentrañar los misterios de la 1H-RMN, comprendiendo qué es un espectro, cómo se interpreta la información crucial del desplazamiento químico, y cómo esta poderosa herramienta nos permite identificar y caracterizar compuestos, desde simples moléculas orgánicas hasta complejos componentes de los alimentos que consumimos.
¿Qué es la RMN de Protón (1H-RMN)?
La Resonancia Magnética Nuclear de Protón, también conocida como hidrógeno-1 RMN o 1H-RMN, es una aplicación de la espectroscopia RMN que se enfoca específicamente en los núcleos de hidrógeno-1 dentro de las moléculas de una sustancia. Prácticamente todo el hidrógeno natural está compuesto por el isótopo 1H, lo que lo convierte en un objetivo ideal para esta técnica. El objetivo principal de la 1H-RMN es determinar la estructura molecular de los compuestos orgánicos.
Un espectro típico de 1H-RMN se caracteriza por señales que aparecen en un rango de desplazamiento químico que va, aproximadamente, de +14 a -4 partes por millón (ppm). Además de la posición de estas señales (el desplazamiento químico), el espectro proporciona información valiosa a través del acoplamiento espín-espín entre los protones y la integración de las señales, que refleja la abundancia relativa de cada tipo de protón en la molécula. Estas tres piezas de información (desplazamiento químico, multiplicidad por acoplamiento y área de la señal) son cruciales para la asignación estructural.
Para obtener un espectro de RMN de alta calidad, es común utilizar solventes deuterados (como CDCl3 o D2O). El deuterio (D o 2H) es un isótopo del hidrógeno que no es activo en RMN de protón, lo que significa que no produce señales en el espectro 1H. Esto permite que las señales de los protones de la muestra se observen sin interferencia del solvente. Además, la señal de deuterio del solvente se utiliza para el bloqueo de frecuencia-campo (deuterium lock), compensando la deriva natural del campo magnético del espectrómetro y asegurando la estabilidad de la señal.
Históricamente, y aún hoy, un compuesto llamado tetrametilsilano (TMS, (CH3)4Si) se utiliza como estándar interno para referenciar los desplazamientos químicos. Los protones del TMS son químicamente equivalentes, produciendo una única señal muy nítida que se define como 0 ppm. Debido a su volatilidad, el TMS facilita la recuperación de la muestra. Los espectrómetros modernos pueden también referenciar los espectros basándose en la señal del protón residual del propio solvente deuterado, ya que la diferencia entre la frecuencia de resonancia del solvente y 0 ppm (TMS) es bien conocida.
El Corazón de la RMN: El Desplazamiento Químico
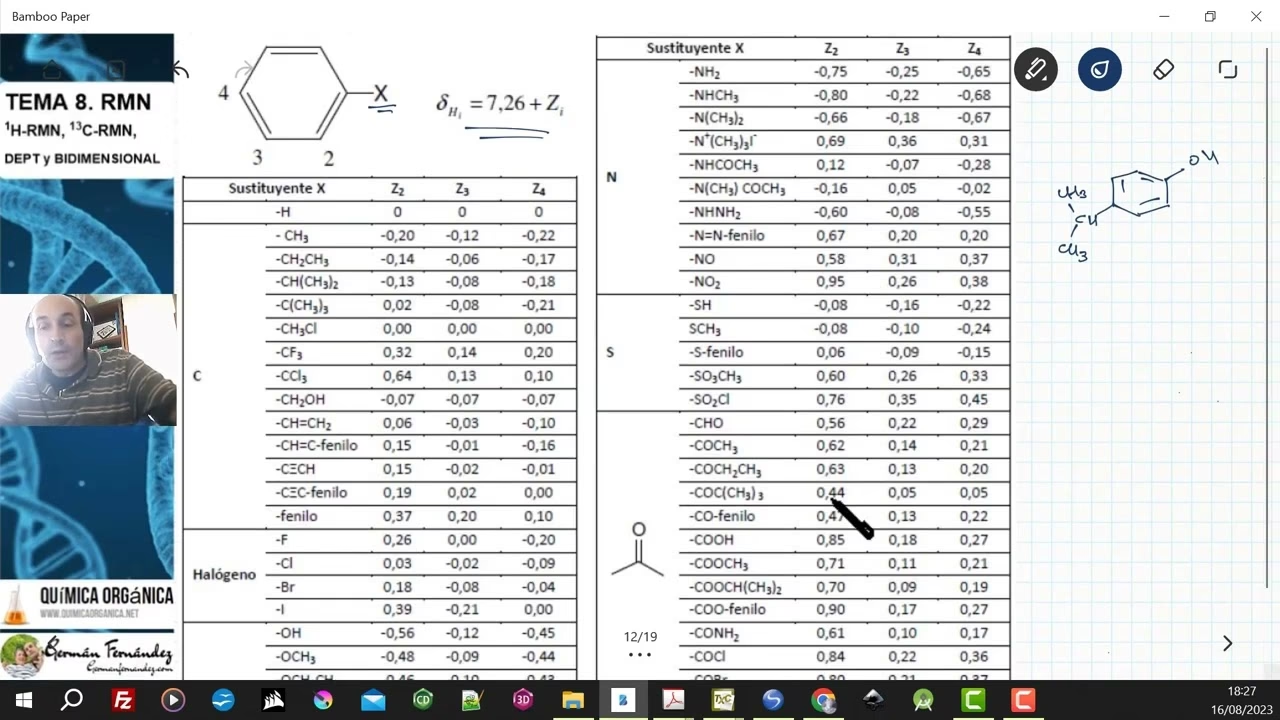
Los desplazamientos químicos en la espectroscopia RMN se refieren al fenómeno por el cual la frecuencia de resonancia de un núcleo en un campo magnético se ve influenciada por su entorno químico. Este efecto surge del apantallamiento o desapantallamiento del núcleo por la nube electrónica circundante. Cuando un núcleo experimenta diferentes densidades electrónicas locales debido a átomos cercanos o grupos funcionales, su frecuencia de resonancia se altera en relación con un estándar de referencia, generalmente el TMS en solventes orgánicos.
Esta alteración se expresa en partes por millón (ppm) y es lo que conocemos como desplazamiento químico (δ). La ecuación fundamental para el desplazamiento químico es:
δ (ppm) = [(Frecuencia de la muestra - Frecuencia de referencia) / Frecuencia de referencia] x 10^6
Donde la frecuencia de referencia es la del TMS. Es decir, el desplazamiento químico no es un valor absoluto, sino una medida relativa de la frecuencia de resonancia de un protón en comparación con el protón del TMS. Un valor de δ más alto (a campo bajo) indica que el protón está más desapantallado, mientras que un valor más bajo (a campo alto) indica que está más apantallado.
La tendencia del desplazamiento químico se ve afectada principalmente por la proximidad a átomos electronegativos y a grupos insaturados:
- Átomos Electronegativos: Grupos como el oxígeno (-O-), nitrógeno (-N-), o los halógenos (F, Cl, Br, I) retiran densidad electrónica de los protones cercanos. Esto reduce el apantallamiento del protón, haciendo que resuene a una frecuencia más alta, es decir, un valor de δ mayor (a campo bajo). Por ejemplo, los protones en un grupo -CH2-O- resonarán a un valor de ppm más alto que los de un -CH2-CH2-. Este efecto disminuye rápidamente con la distancia.
- Grupos Insaturados: Los grupos con enlaces dobles o triples (C=C, C=O) y los anillos aromáticos también causan un desapantallamiento significativo. En el caso de los grupos insaturados, si el núcleo afectado está en el plano de la insaturación, se produce un desplazamiento a campo bajo. Sin embargo, en las regiones por encima y por debajo de este plano, puede ocurrir un apantallamiento. Los protones aromáticos, por ejemplo, están fuertemente desapantallados debido a la corriente de anillo diamagnética inducida por el campo magnético externo.
Es importante recordar que los valores de desplazamiento químico son aproximados y pueden variar ligeramente dependiendo de la estructura molecular específica, el solvente utilizado, la temperatura y la fuerza del campo magnético del espectrómetro. Sin embargo, estos valores son una referencia invaluable para identificar muchos grupos funcionales y entender la conectividad dentro de una molécula.
Interpretando los Desplazamientos Químicos: Rangos Clave
Aunque no es necesario memorizar cada valor exacto, tener una idea aproximada de los rangos de desplazamiento químico para los tipos más comunes de protones simplifica enormemente la interpretación de los espectros de 1H-RMN. A continuación, se presenta una tabla que resume algunos de los desplazamientos químicos comunes:
| Tipo de Hidrógeno | Desplazamiento Químico (ppm) |
|---|---|
| CH3 alifático (RCH3) | 0.9 - 1.0 |
| CH2 alifático (RCH2R) | 1.2 - 1.7 |
| CH alifático (R3CH) | 1.5 - 2.0 |
| Protones cerca de C=O (CH-C=O) | 2.0 - 2.3 |
| Protones cerca de halógenos (CH-X) | 2.3 - 3.0 |
| Protones en aminas (RNH2) | 1 - 3 |
| Protones en metilos aromáticos (ArCH3) | 2.2 - 2.4 |
| Protones en éteres (ROCH3) | 3.7 - 3.9 |
| Protones en alcoholes (ROH) | 1 - 5 (variable) |
| Protones en alquenos (C=CH) | 4.5 - 6.5 |
| Protones aromáticos (ArH) | 6.0 - 8.7 |
| Protones en aldehídos (RCHO) | 9.5 - 10.0 |
| Protones en ácidos carboxílicos (RCOOH) | 10 - 13 |
¿Cómo se desplazan los grupos aromáticos en la RMN?
Una de las características más distintivas de los espectros de 1H-RMN es el desplazamiento de los protones aromáticos. Los protones unidos directamente a anillos aromáticos (Ar-H) típicamente resuenan en el rango de 6.0 a 8.7 ppm, un valor considerablemente a campo bajo. Este desapantallamiento se debe a un fenómeno conocido como la corriente de anillo diamagnética. Cuando un anillo aromático se coloca en un campo magnético externo, los electrones π deslocalizados dentro del anillo circulan, generando un pequeño campo magnético inducido. Este campo inducido se alinea con el campo externo en la región donde se encuentran los protones del anillo, sumándose a él y provocando que los protones experimenten un campo magnético local más fuerte, lo que resulta en un desapantallamiento y, por lo tanto, un desplazamiento a campo bajo.
Por ejemplo, el benceno, una molécula aromática simple, exhibe una única señal nítida alrededor de 7.2 ppm, lo que es un claro indicador de la presencia de protones aromáticos. Otros grupos, como los metilos unidos a anillos aromáticos (Ar-CH3), también pueden presentar desplazamientos ligeramente más altos de lo esperado para un grupo metilo alifático (2.2 – 2.4 ppm) debido a la influencia electrónica del anillo.
Más Allá del Desplazamiento: Acoplamiento Espín-Espín y Multiplicidad
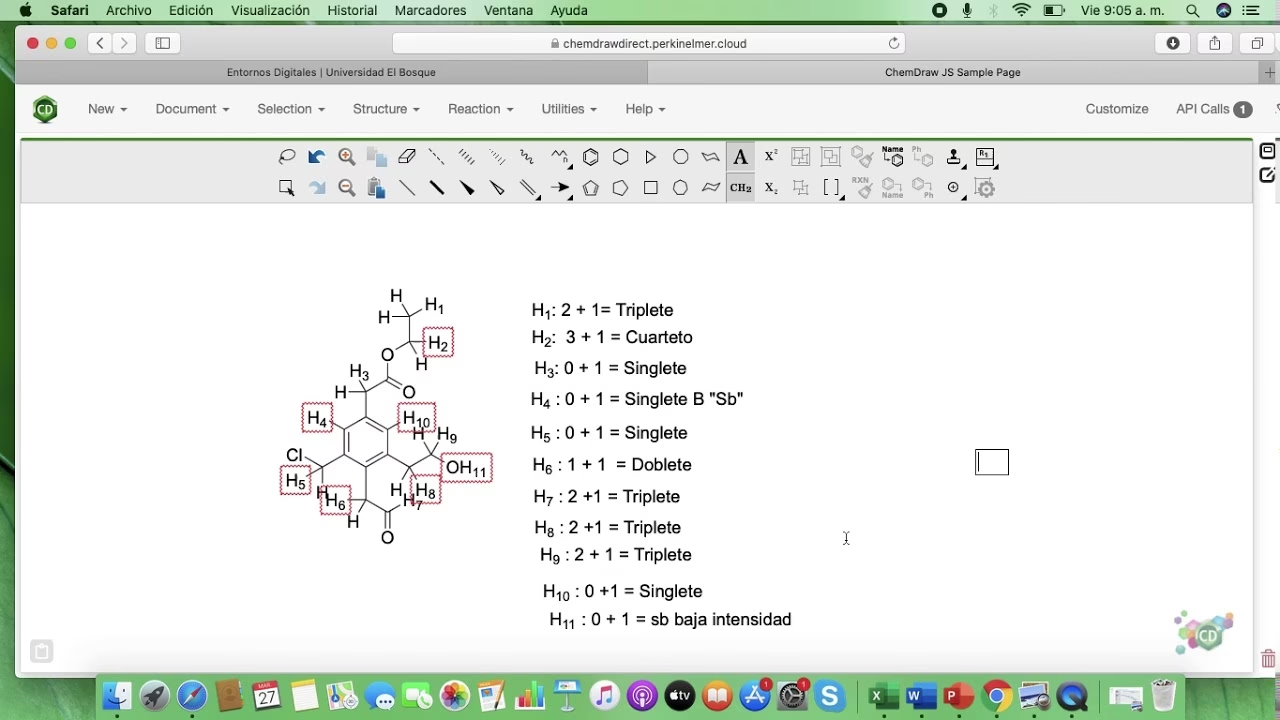
Además del desplazamiento químico, los espectros de RMN permiten asignaciones estructurales gracias al acoplamiento espín-espín. Este fenómeno ocurre porque los núcleos, al poseer un pequeño campo magnético propio, influyen entre sí, cambiando la energía y, por lo tanto, la frecuencia de los núcleos cercanos a medida que resuenan. El tipo más importante en la RMN básica es el acoplamiento escalar, que se produce a través de enlaces químicos y es típicamente visible hasta tres enlaces de distancia (acoplamiento 3-J), aunque ocasionalmente puede verse a distancias de cuatro o cinco enlaces, aunque estos suelen ser considerablemente más débiles.
El efecto del acoplamiento escalar se manifiesta como la división de una señal en múltiples picos, un fenómeno conocido como multiplicidad. La magnitud de esta división se llama constante de acoplamiento y se mide en Hertz (Hz). La constante de acoplamiento es independiente de la fuerza del campo magnético del espectrómetro, ya que es causada por el campo magnético de otro núcleo, no por el imán del espectrómetro.
La regla simple para predecir la multiplicidad de una señal es la regla "n+1": un protón con 'n' protones vecinos equivalentes (a tres enlaces de distancia) aparecerá como un grupo de 'n+1' picos. Las intensidades relativas de estos picos siguen las proporciones del Triángulo de Pascal:
| n (Vecinos) | Nombre | Relación de Intensidades (Triángulo de Pascal) |
|---|---|---|
| 0 | Singlete | 1 |
| 1 | Doble (Dobleto) | 1: 1 |
| 2 | Triple (Triplete) | 1: 2: 1 |
| 3 | Cuartete | 1: 3: 3: 1 |
| 4 | Quintete | 1: 4: 6: 4: 1 |
| 5 | Sextete | 1: 5: 10: 10: 5: 1 |
| 6 | Septete | 1: 6: 15: 20: 15: 6: 1 |
Por ejemplo, el espectro del cloruro de etilo (CH3CH2Cl) muestra un triplete para los protones del CH3 (n=2, por los dos protones del CH2) y un cuartete para los protones del CH2 (n=3, por los tres protones del CH3), con una relación de integración de 3:2, respectivamente.
Cuando un protón está acoplado a dos protones diferentes con constantes de acoplamiento distintas, se observará un doblete de dobletes en lugar de un triplete. La complejidad de estos multipletes puede aumentar significativamente, y su análisis detallado proporciona pistas cruciales sobre la estructura molecular.
Aplicaciones Prácticas de la RMN de 1H
La RMN de 1H es una de las técnicas analíticas más versátiles y potentes disponibles para los químicos, con aplicaciones que van mucho más allá de la simple identificación de compuestos. Su capacidad para proporcionar un perfil global de los protones en una molécula la convierte en una herramienta estructural indispensable.
- Determinación Estructural: Es su aplicación más fundamental. Al combinar el desplazamiento químico (que indica el entorno electrónico), la multiplicidad (que revela la conectividad con protones vecinos) y la integración (que proporciona el número relativo de protones de cada tipo), los químicos pueden reconstruir la estructura tridimensional de una molécula.
- Análisis de Mezclas: Las intensidades integradas de las señales de RMN son, idealmente, proporcionales a la relación de los núcleos dentro de la molécula. Esto significa que para mezclas, las intensidades de las señales pueden utilizarse para determinar las proporciones molares de los componentes, una capacidad vital en el control de calidad y la investigación.
- Identificación de Grupos Funcionales: Los rangos característicos de desplazamiento químico permiten identificar rápidamente la presencia de grupos funcionales como alcoholes, aldehídos, ácidos carboxílicos, éteres, alquenos o anillos aromáticos.
- Estudios de Adulteración y Discriminación: La RMN de 1H ha demostrado ser muy eficaz en el control de calidad de alimentos. Por ejemplo, los espectros de 1H-RMN de varios jugos de frutas (limón, pomelo, naranja, manzana, albaricoque) han sido publicados, y sus señales asignadas a azúcares, aminoácidos, alcoholes, ácidos y polifenoles. Esta información, combinada con técnicas quimiométricas, se utiliza para discriminar entre variedades de manzanas o para detectar la adulteración de jugos de naranja y manzana. A diferencia de otros métodos que requieren estándares específicos de marcadores, la RMN ofrece un perfil global, permitiendo la detección de adulteraciones incluso sin una asignación total de todas las señales.
- Estudio de Cambios Bioquímicos: La RMN de carbono-13 (13C-RMN) en estado sólido, por ejemplo, se ha utilizado para estudiar frutas, permitiendo seguir los cambios en los polímeros de la pared celular durante la maduración.
En resumen, la RMN de 1H no solo nos dice qué protones están presentes, sino también dónde están ubicados en la molécula, a qué otros protones están conectados y en qué cantidad, proporcionando una vista microscópica esencial para la química.
Preguntas Frecuentes (FAQ)
¿Qué es TMS y por qué se usa como referencia en RMN?
TMS (Tetrametilsilano, (CH3)4Si) es un compuesto orgánico que se utiliza como estándar de referencia para definir el 0 ppm en los espectros de RMN. Se elige por varias razones: sus doce protones son químicamente equivalentes, lo que produce una única y nítida señal; es químicamente inerte y no reactivo con la mayoría de las muestras; es volátil, lo que facilita su eliminación de la muestra después del análisis; y sus protones están muy apantallados, resonando a un campo más alto que la mayoría de los otros protones orgánicos, lo que asegura que su señal no se superpondrá con las señales de la muestra.
¿Por qué los protones aromáticos aparecen a campo bajo en el espectro de RMN?
Los protones aromáticos (Ar-H) aparecen a campo bajo (valores de δ más altos, típicamente entre 6.0 y 8.7 ppm) debido al efecto de la corriente de anillo diamagnética. Cuando un anillo aromático se expone a un campo magnético externo, los electrones π deslocalizados dentro del anillo generan una corriente. Esta corriente induce un campo magnético local que se suma al campo externo en la posición de los protones del anillo, haciendo que estos protones experimenten un campo magnético efectivo mayor y, por lo tanto, resuenen a una frecuencia más alta (desapantallamiento).
¿Qué indica la integración de una señal en RMN?
La integración (o el área bajo cada señal o grupo de señales) en un espectro de RMN es directamente proporcional al número de protones que contribuyen a esa señal. Por lo tanto, las relaciones de integración nos dan la relación relativa de los diferentes tipos de protones en la molécula, o en el caso de una mezcla, las proporciones molares de los componentes.
¿Qué es un "D2O shake" y para qué sirve?
Un "D2O shake" (agitación con D2O) es un método experimental en RMN de 1H para identificar protones lábiles, es decir, protones que pueden intercambiarse con el deuterio del agua pesada (D2O). Estos incluyen protones de grupos -OH (alcoholes), -NH2 (aminas) o -SH (tioles), que no tienen un desplazamiento químico característico y a menudo aparecen como picos anchos. Al añadir D2O a la muestra y agitar, el deuterio reemplaza a estos protones lábiles. Como el deuterio no es activo en RMN de 1H, la señal correspondiente al protón lábil desaparece del espectro, confirmando su naturaleza.
¿El desplazamiento químico es un valor exacto?
No, los valores de desplazamiento químico no son exactos, sino más bien valores típicos o de referencia. Las desviaciones pueden ser de ±0.2 ppm o incluso más. El valor exacto de un desplazamiento químico depende de la estructura molecular específica, el solvente utilizado, la temperatura, la fuerza del campo magnético del espectrómetro y la influencia de otros grupos funcionales vecinos. Por lo tanto, deben ser utilizados como guías y no como cifras absolutas.
En conclusión, la RMN de Protón, con su capacidad para revelar el desplazamiento químico, el acoplamiento espín-espín y las integraciones de las señales, es una herramienta indispensable en el arsenal de cualquier químico. Nos permite realizar 'cálculos' y deducciones complejas sobre la estructura molecular, facilitando desde el descubrimiento de nuevos fármacos hasta la detección de adulteraciones en alimentos. Su interpretación requiere una comprensión profunda de cómo el entorno electrónico influye en los núcleos, convirtiendo cada espectro en un rompecabezas molecular esperando ser resuelto.
Si quieres conocer otros artículos parecidos a Desentrañando la RMN de Protón: El Desplazamiento Químico puedes visitar la categoría Cálculos.