04/05/2025
La interacción entre una sustancia y una superficie es un fenómeno fundamental en la química, la física y la biología, con aplicaciones que van desde la catálisis industrial y la purificación de agua hasta el desarrollo de nuevos materiales y fármacos. Esta interacción se conoce como adsorción, un proceso en el que átomos, iones o moléculas de un gas, líquido o sólido disuelto se adhieren a una superficie. La comprensión y, más importante aún, el cálculo preciso de la energía de adsorción son cruciales para predecir el comportamiento de los sistemas y diseñar procesos más eficientes. Históricamente, este cálculo se basaba en modelos empíricos y experimentales, pero la era digital ha abierto las puertas a métodos computacionales avanzados, incluyendo la simulación de alta fidelidad y el revolucionario aprendizaje automático.
En este artículo, exploraremos en profundidad qué es la energía de adsorción, por qué su cálculo es tan vital y cómo ha evolucionado la metodología para determinarla. Recorreremos desde los pilares clásicos como los modelos de Langmuir y Freundlich, que proporcionan una base conceptual sólida, hasta las técnicas de vanguardia basadas en la teoría de funcionales de la densidad (DFT) y el aprendizaje automático, que prometen una precisión y eficiencia sin precedentes en la predicción de estas energías críticas.
- ¿Qué es la Energía de Adsorción y por qué es Importante?
- Métodos Clásicos de Cálculo: Las Isotermas de Adsorción
- Avances en el Cálculo de la Energía de Adsorción: La Revolución Computacional y el Aprendizaje Automático
- Consideraciones Termodinámicas en la Adsorción
- Preguntas Frecuentes sobre la Energía de Adsorción
- Conclusión
¿Qué es la Energía de Adsorción y por qué es Importante?
La energía de adsorción es una medida cuantitativa de la fuerza de interacción entre un adsorbato (la sustancia que se adhiere) y un adsorbente (la superficie a la que se adhiere). Representa la cantidad de energía liberada o absorbida cuando una molécula se une a una superficie. Un valor negativo indica un proceso exotérmico (energía liberada), lo que es común en la mayoría de los procesos de adsorción espontáneos, mientras que un valor positivo implicaría un proceso endotérmico (energía absorbida), que es menos frecuente y requiere un aporte de energía externo.
La importancia de esta energía radica en su capacidad para determinar:
- La estabilidad de la unión: Una mayor energía de adsorción (más negativa) implica una unión más fuerte y estable entre el adsorbato y la superficie.
- La selectividad de un adsorbente: En sistemas con múltiples especies, las diferencias en las energías de adsorción dictan qué moléculas se adsorberán preferentemente.
- La cinética de los procesos: La energía de adsorción influye en la velocidad a la que ocurren la adsorción y la desorción, parámetros clave en procesos catalíticos.
- El diseño de materiales: Permite a los científicos e ingenieros diseñar materiales con propiedades de adsorción específicas para aplicaciones como la separación de gases, la eliminación de contaminantes, el almacenamiento de energía y el desarrollo de sensores.
En esencia, predecir con precisión la energía de adsorción es fundamental para optimizar y desarrollar nuevas tecnologías en una amplia gama de campos científicos e industriales.
Métodos Clásicos de Cálculo: Las Isotermas de Adsorción
Las isotermas de adsorción son representaciones gráficas que describen la cantidad de adsorbato que se adhiere a la superficie de un adsorbente a una temperatura constante, en función de la presión del gas o la concentración del soluto en el equilibrio. Han sido herramientas fundamentales para entender el fenómeno de la adsorción y estimar parámetros clave, incluyendo la capacidad de adsorción y la afinidad.
El Modelo de Adsorción de Langmuir
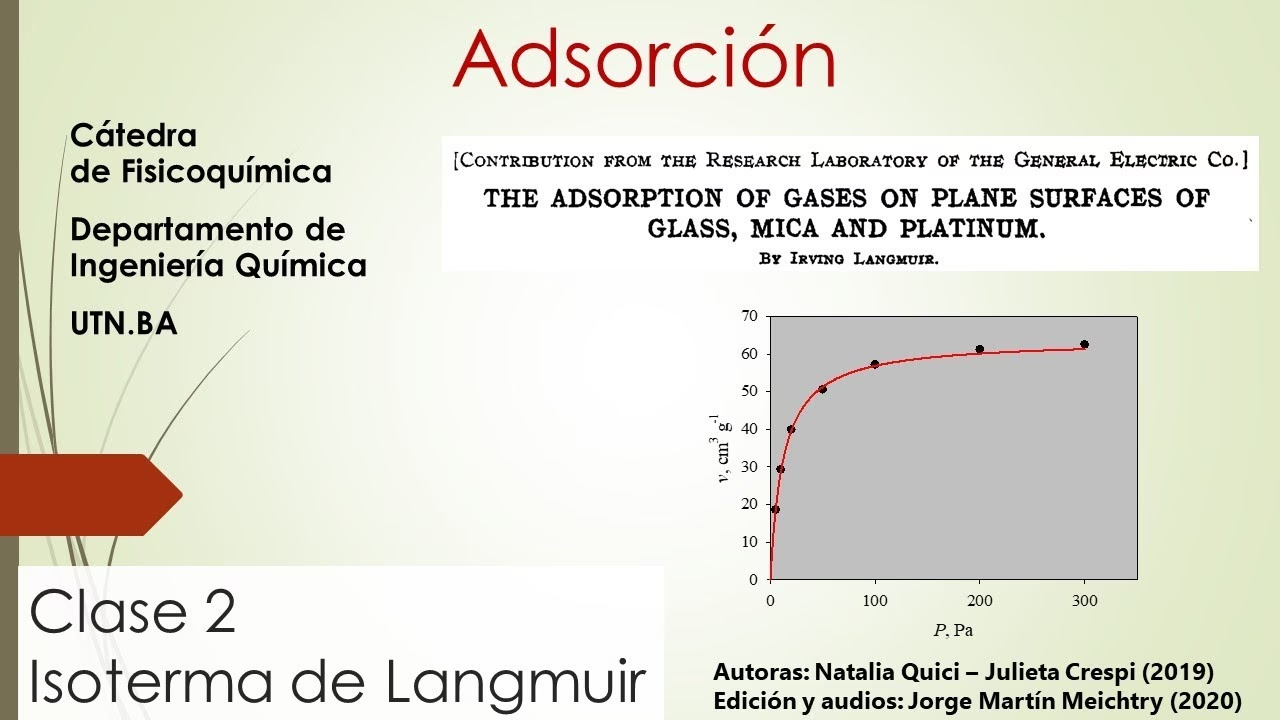
Propuesto por Irving Langmuir en 1916, el modelo de Langmuir es uno de los más sencillos y ampliamente utilizados para describir la adsorción. Se basa en varias suposiciones clave que simplifican la complejidad de las interacciones superficie-molécula:
- La superficie del adsorbente es completamente homogénea, es decir, todos los sitios homogéneos de adsorción son idénticos y tienen la misma energía de adsorción.
- Cada sitio de adsorción puede unirse a un máximo de una molécula de adsorbato (formación de una monocapa).
- No hay interacciones laterales entre las moléculas de adsorbato adyacentes en la superficie.
- La adsorción es reversible, y se alcanza un equilibrio dinámico donde la velocidad de adsorción es igual a la velocidad de desorción.
Fórmula para la Cobertura de Superficie de Langmuir (θA)
La ecuación de Langmuir relaciona la fracción de la superficie cubierta (θA) con la presión parcial del gas (pA) o la concentración del soluto (CA) en el equilibrio. Para la adsorción en fase gaseosa, la cobertura de la superficie θA se expresa como:
θA = (KeqA * pA) / (1 + KeqA * pA)
Donde:
- θA es la fracción de sitios de adsorción ocupados en la superficie (cobertura).
- pA es la presión parcial del adsorbato A en el equilibrio (para gases).
- KeqA (o KL) es la constante de equilibrio de adsorción de Langmuir, que refleja la afinidad del adsorbato por el adsorbente.
Para soluciones, pA se reemplaza por CA (concentración del adsorbato en el equilibrio).
¿Qué es KL en Langmuir?
La constante KL (o KeqA en la notación más general) en el contexto de la isoterma de Langmuir es un parámetro crucial que representa la afinidad del adsorbato por el adsorbente y la capacidad de enlace. Se conoce como la constante de Langmuir o constante de equilibrio de adsorción. Sus unidades pueden variar (por ejemplo, L/mg o L/mol) dependiendo de si se trabaja con concentración o presión. Un valor más alto de KL indica una mayor afinidad, lo que significa que el adsorbato se adsorberá más fácilmente en la superficie. Matemáticamente, KL se obtiene de la linealización de la ecuación de Langmuir, generalmente graficando Ce/qe versus Ce, donde 1/(qmaxKL) es la pendiente y 1/qmax es la ordenada al origen, siendo qe la cantidad adsorbida en el equilibrio y qmax la capacidad máxima de adsorción.

Limitaciones del Modelo de Langmuir
A pesar de su simplicidad y utilidad, el modelo de Langmuir tiene limitaciones significativas debido a sus suposiciones idealizadas:
- Superficies heterogéneas: La mayoría de las superficies reales no son perfectamente homogéneas; tienen sitios con diferentes energías de adsorción (rugosidad, defectos).
- Interacciones adsorbato-adsorbato: Las moléculas adsorbidas a menudo interactúan entre sí (atractiva o repulsivamente), lo que afecta la energía de adsorción de moléculas adyacentes y no está contemplado en el modelo básico.
- Adsorción multicapa: Langmuir asume la formación de una monocapa, pero en muchos casos, especialmente a altas presiones, pueden formarse múltiples capas de adsorbato.
El Modelo de Adsorción de Freundlich
El modelo de isoterma de Freundlich, propuesto por Herbert Freundlich en 1906, es un modelo empírico que describe la adsorción en superficies heterogéneas y permite la formación de multicapas. Su ecuación es:
qe = KF * Cen
Donde:
- qe es la cantidad de adsorbato adsorbido por unidad de masa del adsorbente en el equilibrio.
- Ce es la concentración del adsorbato en la fase líquida en el equilibrio.
- KF es la constante de Freundlich, un indicador de la capacidad de adsorción del adsorbente.
- n es la constante de intensidad de adsorción, que representa la heterogeneidad de la superficie y la favorabilidad del proceso. Un valor de n entre 2 y 10 generalmente indica una buena intensidad de adsorción, y valores cercanos a 1 sugieren sitios energéticamente equivalentes.
A diferencia de Langmuir, Freundlich no asume una capacidad máxima de adsorción ni sitios homogéneos, lo que a menudo lo hace más adecuado para sistemas complejos.
Tabla Comparativa: Langmuir vs. Freundlich
Para ilustrar las diferencias clave entre estos dos modelos clásicos, presentamos la siguiente tabla comparativa:
| Característica | Modelo de Langmuir | Modelo de Freundlich |
|---|---|---|
| Tipo de superficie | Homogénea (sitios idénticos) | Heterogénea (sitios con diferentes energías) |
| Capacidad de adsorción | Monocapa (capacidad máxima finita) | Multicapa posible (no hay capacidad máxima definida) |
| Interacciones laterales | No hay interacciones | No consideradas explícitamente, pero el modelo las acomoda implícitamente |
| Base | Teórica (suposiciones cinéticas/termodinámicas) | Empírica |
| Aplicabilidad | Adsorción quimisorción a bajas concentraciones | Adsorción fisisorción y quimisorción en un amplio rango de concentraciones, superficies heterogéneas |
| Constantes | KL (KeqA), qmax | KF, n |
Avances en el Cálculo de la Energía de Adsorción: La Revolución Computacional y el Aprendizaje Automático
A pesar de la utilidad de los modelos clásicos, su naturaleza simplificada limita su precisión y predictibilidad en sistemas complejos del mundo real. La necesidad de una comprensión más profunda y una predicción más precisa de la energía de adsorción ha impulsado el desarrollo de métodos computacionales avanzados.
El Enfoque de la Teoría de Funcionales de la Densidad (DFT)
La teoría de funcionales de la densidad (DFT) se ha convertido en el estándar de oro para el cálculo ab initio de las energías de adsorción. La DFT es un método de mecánica cuántica que permite calcular la estructura electrónica (y, por ende, las propiedades) de sistemas de muchos cuerpos, como moléculas y sólidos. Para la adsorción, la energía de adsorción se calcula como la diferencia de energía total entre el sistema adsorbate-superficie, la superficie aislada y el adsorbato aislado, todo ello en sus configuraciones de mínima energía.
Aunque la DFT proporciona una aproximación muy cercana a la "verdad fundamental" (ground truth) de la energía de adsorción, es computacionalmente muy costosa, especialmente para explorar el vasto espacio de configuraciones posibles de un adsorbato en una superficie (orientaciones, posiciones). El número de configuraciones es combinatorio, lo que hace inviable explorar todas las posibilidades mediante DFT pura.
El Dataset OC20-Dense: Una Base para la Innovación
Para abordar el desafío computacional y mejorar la evaluación de los métodos de cálculo de la energía de adsorción, se desarrolló el dataset Open Catalyst 2020-Dense (OC20-Dense). Este dataset es fundamental porque aproxima la energía de adsorción verdadera explorando densamente numerosas configuraciones para cada sistema único adsorbato-superficie. OC20-Dense contiene alrededor de 1000 combinaciones únicas de adsorbato-superficie, que abarcan 74 adsorbatos y más de 800 estructuras cristalinas inorgánicas, sumando más de 80,000 configuraciones generadas heurística y aleatoriamente. La creación de este dataset requirió aproximadamente 4 millones de horas de CPU, lo que subraya la magnitud del esfuerzo computacional necesario para mapear este espacio de configuraciones.
Este dataset permite evaluar modelos de aprendizaje automático frente a una estimación más precisa de la energía de adsorción mínima global, en lugar de depender únicamente de configuraciones heurísticas (conocidas como DFT-Heuristic-Only o DFT-Heur), que pueden pasar por alto configuraciones de menor energía.
Redes Neuronales Gráficas (GNNs) y su Potencial
Las Redes Neuronales Gráficas (GNNs, por sus siglas en inglés) han demostrado ser herramientas excepcionalmente poderosas en el campo de la química computacional y los materiales. Modelos como SchNet, DimeNet++, PaiNN, GemNet-OC y SCN-MD-Large han sido utilizados y evaluados en datasets como OC20 y OC20-Dense para predecir propiedades moleculares y de materiales, incluyendo las energías de adsorción. Estas GNNs pueden aprender las relaciones complejas entre la estructura atómica y las propiedades energéticas, lo que les permite hacer predicciones rápidas una vez entrenadas. Sin embargo, incluso los modelos de GNN más avanzados no siempre logran la precisión de la DFT, especialmente en la relajación de estructuras a sus verdaderos mínimos de energía.

Estrategias Híbridas ML+DFT: El Algoritmo AdsorbML
Dado que las predicciones de ML son rápidas pero pueden carecer de la precisión de la DFT para encontrar el mínimo de energía global, y la DFT es precisa pero lenta, la combinación de ambos enfoques es una solución prometedora. Aquí es donde entra el algoritmo AdsorbML.
El algoritmo AdsorbML utiliza el aprendizaje automático para acelerar el proceso de colocación del adsorbato, seguido de cálculos DFT para refinar la determinación de la energía de adsorción. La estrategia general implica generar configuraciones iniciales (heurísticas y aleatorias), realizar relajaciones rápidas con ML, clasificar los sistemas por su energía ML, y luego pasar las 'k' mejores configuraciones a DFT para un cálculo de alta precisión.
AdsorbML con Evaluaciones de Punto Único (ML+SP)
En esta estrategia, después de que las relajaciones ML han clasificado las configuraciones de menor a mayor energía, los 'k' sistemas con las energías más bajas se someten a cálculos DFT de punto único (SP). Esto significa que la estructura relajada por ML se congela, y la DFT simplemente calcula su energía en esa configuración. Esta aproximación es extremadamente rápida y ofrece mejoras significativas en la velocidad de cálculo.
- Resultados: Modelos como eSCN-MD-Large y GemNet-OC-MD-Large lograron tasas de éxito superiores al 86% para k=5, con eSCN-MD-Large alcanzando un 88.27% (ligeramente mejor que la línea base DFT-Heur). Las aceleraciones son impresionantes, oscilando entre 1384x y 1388x para k=5, y hasta 6817x para k=1 con tasas de éxito aún aceptables (82% para eSCN-MD-Large).
AdsorbML con Relajaciones Completas (ML+RX)
La segunda estrategia implica que, después de la relajación ML y la selección de los 'k' mejores sistemas, se realizan relajaciones completas (RX) utilizando DFT a partir de esas estructuras pre-optimizadas por ML. Esto es más costoso computacionalmente que ML+SP, pero tiene el potencial de refinar aún más la estructura y encontrar mínimos de energía más profundos, ya que la DFT completa el proceso de optimización.
- Resultados: eSCN-MD-Large y GemNet-OC-MD-Large superaron a todos los modelos con tasas de éxito del 90.60% y 91.61% para k=5, respectivamente. Las aceleraciones son menores en comparación con ML+SP (215x y 172x para k=5), debido al mayor costo computacional de las relajaciones DFT completas. Sin embargo, estas aceleraciones siguen siendo sustanciales en comparación con un enfoque puramente DFT.
Comparativa de Estrategias: Precisión vs. Eficiencia
La elección entre ML+SP y ML+RX depende del equilibrio deseado entre precisión y eficiencia. Los resultados sugieren que ML+SP es casi 8 veces más rápido que ML+RX con solo una disminución marginal del rendimiento (3-4% en las tasas de éxito) para los mejores modelos. Esto hace que ML+SP sea una opción atractiva si la velocidad es la prioridad principal y una ligera reducción en la precisión es aceptable. Para modelos menos precisos en la relajación (como SchNet o DimeNet++), ML+RX es más beneficioso, ya que la relajación DFT puede corregir las estructuras subóptimas predichas por el ML.
Análisis de la Configuración y Generalización
La efectividad de AdsorbML también fue evaluada en diferentes subdivisiones del dataset OC20-Dense (ID, OOD-Adsorbate, OOD-Catalyst, OOD-Both), lo que significa que se probó la capacidad de generalización del modelo a adsorbatos y superficies que no estaban en el conjunto de entrenamiento. Los resultados mostraron que el rendimiento se mantuvo consistente en estas subdivisiones, lo que indica que AdsorbML es robusto y puede proporcionar resultados precisos y significativos incluso para aplicaciones con adsorbatos o superficies fuera del dominio de entrenamiento inicial. Además, se encontró que las configuraciones generadas aleatoriamente tienen un impacto significativo en la tasa de éxito, incluso mayor que las heurísticas, lo que subraya la importancia de explorar un espacio de configuración diverso.
Consideraciones Termodinámicas en la Adsorción
Más allá de las isotermas y los cálculos de energía, la termodinámica proporciona un marco fundamental para comprender la espontaneidad y la naturaleza de la adsorción. Los cambios en la entalpía (ΔH), entropía (ΔS) y energía libre de Gibbs (ΔG) son cruciales:
- ΔH (Entalpía): Un ΔH negativo indica que la adsorción es un proceso exotérmico (libera calor), lo cual es común. Un ΔH positivo (endotérmico) es menos frecuente y sugiere que el calor es necesario para que ocurra la adsorción.
- ΔS (Entropía): Un ΔS negativo para la adsorción (disminución de la aleatoriedad en la interfaz) es típico, ya que las moléculas pasan de un estado más libre (gas o solución) a un estado más ordenado en la superficie.
- ΔG (Energía Libre de Gibbs): La espontaneidad de un proceso se rige por el cambio en la energía libre de Gibbs (ΔG = ΔH - TΔS). Un ΔG negativo indica un proceso espontáneo. Para la adsorción, valores de ΔG negativos que aumentan con la temperatura sugieren que el proceso es menos espontáneo a temperaturas más altas, lo cual es consistente con la naturaleza exotérmica de la adsorción (donde un aumento de temperatura favorece la desorción).
Estos parámetros termodinámicos son esenciales para el diseño y escalado de procesos de adsorción, ya que permiten predecir cómo variarán las propiedades de adsorción con la temperatura y otras condiciones.
Preguntas Frecuentes sobre la Energía de Adsorción
¿Para qué se usa la energía de adsorción?
La energía de adsorción se utiliza para comprender la fuerza de unión entre moléculas y superficies, lo que es vital en el diseño de catalizadores (para acelerar reacciones), materiales para purificación de agua o aire, almacenamiento de gases (como hidrógeno o CO2), y en la fabricación de sensores y recubrimientos protectores.
¿Cuál es la diferencia entre fisisorción y quimisorción?
La fisisorción (adsorción física) implica fuerzas débiles de Van der Waals (baja energía de adsorción, reversible, no forma enlaces químicos). La quimisorción (adsorción química) implica la formación de enlaces químicos fuertes entre el adsorbato y la superficie (alta energía de adsorción, a menudo irreversible, específica).
¿Por qué los modelos ML+DFT son mejores que solo ML o solo DFT?
Los modelos híbridos ML+DFT combinan la velocidad y eficiencia del aprendizaje automático para explorar un gran número de configuraciones y pre-optimizar estructuras, con la alta precisión de la teoría de funcionales de la densidad (DFT) para refinar las energías de adsorción. Esto permite obtener resultados precisos en una fracción del tiempo que tomaría un cálculo puramente DFT, superando las limitaciones de ambos enfoques por separado.
¿Es siempre el modelo de Langmuir el mejor?
No, el modelo de Langmuir es ideal para superficies homogéneas con adsorción en monocapa y sin interacciones entre moléculas adsorbidas. Para superficies heterogéneas o cuando hay interacciones significativas o formación de multicapas, modelos como Freundlich o BET pueden ser más adecuados. La elección del modelo depende de la naturaleza del sistema experimental.
¿Qué significa un valor de 'n' en la isoterma de Freundlich?
En la isoterma de Freundlich, 'n' es la constante de intensidad de adsorción. Si n > 1, la adsorción es favorable. Valores de 'n' entre 2 y 10 indican una buena intensidad de adsorción, mientras que valores cercanos a 1 sugieren que los sitios activos del adsorbente son energéticamente equivalentes.
Conclusión
El cálculo de la energía de adsorción es un pilar fundamental en la ciencia de superficies y materiales. Desde los modelos empíricos y simplificados de Langmuir y Freundlich, que sentaron las bases para nuestra comprensión inicial, hasta las sofisticadas técnicas computacionales basadas en la teoría de funcionales de la densidad (DFT), hemos sido testigos de una evolución notable en la precisión y eficiencia de estos cálculos. La irrupción del aprendizaje automático, especialmente en combinación con la DFT a través de estrategias híbridas como AdsorbML, ha revolucionado la capacidad de explorar espacios de configuración vastos y complejos, permitiendo predicciones de energía de adsorción con una velocidad y fiabilidad sin precedentes. Estos avances no solo profundizan nuestra comprensión de las interacciones moleculares en las superficies, sino que también aceleran drásticamente el descubrimiento y diseño de nuevos materiales con propiedades optimizadas para los desafíos tecnológicos y ambientales del futuro.
Si quieres conocer otros artículos parecidos a Desentrañando la Energía de Adsorción: Métodos y Avances puedes visitar la categoría Cálculos.