18/07/2022
Las proteínas son las máquinas moleculares de la vida, desempeñando roles fundamentales en prácticamente todos los procesos biológicos, desde la catálisis de reacciones químicas hasta la señalización celular y el transporte de moléculas. Dada su omnipresencia y su importancia funcional, la capacidad de detectar, cuantificar y analizar proteínas es una piedra angular de la investigación en biología, biomedicina y biotecnología. Medir con precisión la concentración de proteínas en una muestra es un paso esencial en innumerables aplicaciones, que van desde la purificación y el marcaje de proteínas para experimentos subsiguientes hasta la preparación de muestras para técnicas analíticas más complejas como la electroforesis o la espectrometría de masas. Comprender la cantidad de proteína presente es crucial para asegurar la reproducibilidad de los experimentos, optimizar las condiciones de reacción y obtener resultados significativos.
Este artículo explorará en profundidad los diversos métodos disponibles para la cuantificación de proteínas, detallando sus principios, ventajas, limitaciones y aplicaciones específicas. Además, abordaremos cómo estas técnicas se integran con métodos de análisis más amplios, como la electroforesis en gel, para proporcionar una comprensión integral de las proteínas en muestras biológicas.
- ¿Por qué es crucial medir la concentración de proteínas?
- Métodos de Cuantificación de Proteínas: Un Abanico de Opciones
- Absorbancia a 280 nm: El Método Directo y Rápido
- Métodos Colorimétricos: Mayor Sensibilidad y Amplia Aplicación
- Métodos Fluorimétricos: La Cúspide de la Sensibilidad
- Detección y Análisis de Proteínas Específicas: El Ensayo ELISA
- El Uso de Proteínas Fluorescentes en el Análisis Biológico
- Instrumentación para la Cuantificación y Análisis de Proteínas: Los Lectores de Microplacas
- La Electroforesis en Gel: Una Herramienta Fundamental para el Análisis de Proteínas
- Preguntas Frecuentes sobre la Cuantificación y Análisis de Proteínas
- ¿Cuál es el método más sensible para cuantificar proteínas?
- ¿Se puede usar la electroforesis para cuantificar proteínas?
- ¿Qué es el TEMED y el APS y para qué sirven en el laboratorio?
- ¿Cómo se determina el tamaño de una proteína en un gel de electroforesis?
- ¿Qué factores influyen en la migración de las proteínas en un gel de SDS-PAGE?
¿Por qué es crucial medir la concentración de proteínas?
La medición precisa de la concentración de proteína total en una muestra no es solo una buena práctica de laboratorio, sino una necesidad fundamental para el éxito de muchos experimentos. Por ejemplo, al purificar una proteína de interés a partir de un lisado celular, conocer su concentración en cada paso de purificación permite evaluar la eficiencia del proceso y determinar el rendimiento final. En el marcaje de proteínas con fluorocromos o isótopos, una concentración conocida es vital para asegurar una relación molar adecuada entre el marcador y la proteína, garantizando una señal detectable y minimizando el marcaje inespecífico. De manera similar, para preparar muestras destinadas a la electroforesis, como la SDS-PAGE, es imperativo cargar una cantidad consistente de proteína en cada carril del gel para poder realizar comparaciones válidas entre diferentes muestras o condiciones experimentales. Una cuantificación inexacta podría llevar a interpretaciones erróneas de los resultados, desperdicio de reactivos valiosos y una pérdida considerable de tiempo y esfuerzo.
Métodos de Cuantificación de Proteínas: Un Abanico de Opciones
La cuantificación de proteínas se puede abordar de diversas maneras, cada una con sus propias características de sensibilidad, rango dinámico y compatibilidad con diferentes tipos de muestras. Los métodos se dividen generalmente en directos e indirectos, y pueden ser colorimétricos o fluorimétricos.
Absorbancia a 280 nm: El Método Directo y Rápido
Uno de los métodos más rápidos y sencillos para cuantificar proteínas es la medición directa de la absorbancia a 280 nanómetros (nm). Este método se basa en la propiedad de los aminoácidos aromáticos (triptófano, tirosina y, en menor medida, fenilalanina) de absorber la luz ultravioleta en esta longitud de onda. Dada que la mayoría de las proteínas contienen estos aminoácidos, su presencia en una solución se correlaciona directamente con la absorbancia a 280 nm. Se utiliza la ley de Beer-Lambert (A = εbc) para calcular la concentración, donde A es la absorbancia, ε es el coeficiente de extinción molar, b es la longitud de la trayectoria de la luz y c es la concentración.
Ventajas: Es no destructivo, no requiere reactivos adicionales ni incubación, y los resultados son inmediatos. Es ideal para monitorear la concentración durante la purificación de proteínas. Desventajas: Su principal limitación es que el coeficiente de extinción molar (ε) varía significativamente entre diferentes proteínas debido a las diferencias en su composición de aminoácidos aromáticos. Esto significa que la absorbancia a 280 nm de 1 mg/mL de una proteína puede ser diferente de la de 1 mg/mL de otra. Además, la presencia de ácidos nucleicos (ADN o ARN) en la muestra, que también absorben fuertemente a 260 nm y en menor medida a 280 nm, puede interferir y sobreestimar la concentración de proteína. Por lo tanto, este método es más adecuado para muestras de proteínas purificadas y libres de contaminantes de ácidos nucleicos.
Métodos Colorimétricos: Mayor Sensibilidad y Amplia Aplicación
Los métodos colorimétricos son ampliamente utilizados debido a su mayor sensibilidad en comparación con la absorbancia directa a 280 nm y su compatibilidad con una gama más amplia de muestras. Estos métodos implican una reacción química entre la proteína y un reactivo colorimétrico, produciendo un producto que absorbe la luz en el rango visible del espectro, lo que permite su cuantificación mediante un espectrofotómetro o un lector de microplacas.
Ensayo de Ácido Bicinchonínico (BCA)
El ensayo BCA es uno de los métodos colorimétricos más populares. Se basa en dos reacciones: primero, la reducción de iones cúpricos (Cu2+) a iones cuprosos (Cu1+) por los enlaces peptídicos de la proteína en un ambiente alcalino (reacción de Biuret). Luego, el ácido bicinchonínico (BCA) forma un complejo de color púrpura intenso con los iones Cu1+, con una absorbancia máxima a 562 nm. La intensidad del color es directamente proporcional a la concentración de proteína.
Ventajas: Es muy sensible, tiene una buena linealidad en un amplio rango de concentración y es relativamente compatible con muchos detergentes y sales comúnmente utilizados en los laboratorios de bioquímica. Además, el color formado es bastante estable, lo que permite mediciones flexibles. Desventajas: Es susceptible a la interferencia de agentes reductores (como DTT o 2-mercaptoetanol) y agentes quelantes, que pueden reducir el Cu2+ y dar falsos positivos. También requiere un tiempo de incubación a temperatura elevada (generalmente 37°C) para que la reacción se complete.
Ensayo de Bradford
El ensayo de Bradford, o ensayo de Coomassie, es otro método colorimétrico muy utilizado. Se basa en la unión del colorante Coomassie Brilliant Blue G-250 a las proteínas, lo que provoca un cambio en la absorción máxima del colorante. En solución ácida, el Coomassie existe en tres formas: catiónica (rojo), neutra (verde) y aniónica (azul). Cuando el colorante se une a los residuos básicos y aromáticos de las proteínas, se estabiliza la forma aniónica, produciendo un color azul intenso con una absorbancia máxima a 595 nm. La cantidad de color azul es directamente proporcional a la concentración de proteína.
Ventajas: Es muy rápido, económico y compatible con muchos tampones salinos. No es sensible a la mayoría de los agentes reductores o agentes quelantes. Desventajas: Es altamente susceptible a la interferencia de detergentes, especialmente SDS, que pueden unirse al colorante y afectar la medición. También muestra una variabilidad significativa entre diferentes proteínas, ya que la unión del colorante depende de la composición de aminoácidos de la proteína, lo que puede requerir el uso de una curva estándar generada con una proteína similar a la de interés.
Métodos Fluorimétricos: La Cúspide de la Sensibilidad
Para aplicaciones que requieren una sensibilidad excepcional, los métodos fluorimétricos son la opción preferida. Estos métodos utilizan colorantes fluorescentes que se unen específicamente a las proteínas, y la intensidad de la fluorescencia emitida es directamente proporcional a la concentración de proteína. Un ejemplo común es el uso de ensayos basados en fluorocromos como el Qubit (Invitrogen) o NanoOrange (Thermo Fisher Scientific).
Ventajas: Ofrecen una sensibilidad significativamente mayor que los métodos colorimétricos, permitiendo la cuantificación de proteínas en el rango de nanogramos (ng) o incluso picogramos (pg). Tienen un rango dinámico muy amplio y son menos susceptibles a la interferencia de contaminantes comunes como sales, detergentes o ácidos nucleicos, lo que los hace ideales para muestras biológicas complejas y diluidas. Desventajas: Generalmente son más costosos y requieren un fluorímetro o un lector de microplacas con capacidad de detección de fluorescencia.
| Método | Principio | Ventajas | Desventajas | Rango de Sensibilidad Típico |
|---|---|---|---|---|
| Absorbancia 280 nm | Absorción UV por aminoácidos aromáticos (Trp, Tyr) | Rápido, no destructivo, sin reactivos | Interferencia de ácidos nucleicos, variabilidad entre proteínas | 0.1 - 10 mg/mL |
| Bradford | Unión del colorante Coomassie a proteínas (cambio de color) | Rápido, económico, no sensible a reductores | Incompatible con detergentes, variabilidad entre proteínas | 1 - 100 µg/mL |
| BCA | Reducción de Cu2+ a Cu1+ por enlaces peptídicos, formación de complejo BCA-Cu1+ púrpura | Sensible, compatible con detergentes, color estable | Interferencia de agentes reductores, requiere incubación | 0.02 - 2 mg/mL |
| Fluorimétricos (ej. Qubit) | Unión de colorantes fluorescentes específicos a proteínas | Muy alta sensibilidad, amplio rango dinámico, baja interferencia | Más costoso, requiere fluorímetro específico | 0.0001 - 1 µg/mL |
| ELISA | Detección inmunológica de proteínas específicas | Muy alta especificidad y sensibilidad, cuantificación de proteína individual | Requiere anticuerpos específicos, multi-paso, más complejo | pg/mL a ng/mL (dependiendo del ensayo) |
Detección y Análisis de Proteínas Específicas: El Ensayo ELISA
Mientras que los métodos anteriores cuantifican la concentración total de proteína en una muestra, a menudo es necesario identificar y medir la concentración de una proteína específica dentro de una mezcla compleja, como suero sanguíneo, lisados celulares o sobrenadantes de cultivo. Para este propósito, el ensayo de inmunoadsorción ligado a enzima (ELISA) es una técnica invaluable.
El ELISA se basa en la alta especificidad de la interacción antígeno-anticuerpo. En un formato típico, un anticuerpo específico para la proteína de interés se inmoviliza en una microplaca. La muestra se añade, permitiendo que la proteína objetivo se una al anticuerpo inmovilizado. Después de lavar las proteínas no unidas, se añade un segundo anticuerpo (detectable, a menudo marcado con una enzima como la peroxidasa de rábano picante o la fosfatasa alcalina) que se une a la proteína objetivo. Finalmente, se añade un sustrato para la enzima, que se convierte en un producto coloreado o luminiscente, cuya intensidad es proporcional a la cantidad de proteína específica presente. El ELISA es fundamental para el diagnóstico clínico, la investigación de biomarcadores y el estudio de vías de señalización.
El Uso de Proteínas Fluorescentes en el Análisis Biológico
Más allá de la cuantificación de proteínas, el análisis de su localización, dinámica y cambios conformacionales en células vivas ha revolucionado la biología celular. Las proteínas fluorescentes, como la proteína fluorescente verde (GFP) y sus variantes (YFP, CFP, RFP, etc.), han sido herramientas clave para este propósito. Al fusionar genéticamente una proteína de interés con una GFP, los investigadores pueden expresar esta proteína quimérica en células vivas y visualizar su comportamiento en tiempo real bajo un microscopio de fluorescencia. Esto permite estudiar la localización subcelular de las proteínas, su movimiento, interacciones con otras moléculas y cambios en respuesta a estímulos externos, proporcionando una visión dinámica de los procesos biológicos.
Además de las proteínas fluorescentes exógenas, la fluorescencia intrínseca del aminoácido triptófano también se ha utilizado para obtener información valiosa. Las propiedades de emisión de fluorescencia del triptófano son altamente sensibles a su microambiente. Cambios en el plegamiento de una proteína, la unión a ligandos o la interacción con otras moléculas pueden alterar el entorno del triptófano, provocando cambios detectables en su espectro de emisión de fluorescencia. Esto permite a los científicos monitorear los cambios en el estado conformacional de las proteínas, lo cual es crucial para comprender su función y la patogénesis de enfermedades relacionadas con el mal plegamiento proteico.
Instrumentación para la Cuantificación y Análisis de Proteínas: Los Lectores de Microplacas
La eficiencia y la capacidad de alto rendimiento en la cuantificación y el estudio de proteínas han sido posibles gracias al desarrollo de instrumentos especializados, en particular los lectores de microplacas. Estos dispositivos versátiles pueden medir la absorbancia, la fluorescencia y la luminiscencia en múltiples pocillos de una placa (generalmente de 96 o 384 pocillos) de forma rápida y automatizada. Esto es invaluable para los ensayos colorimétricos (como BCA y Bradford), fluorimétricos y ELISA, permitiendo a los investigadores procesar un gran número de muestras simultáneamente y obtener resultados consistentes y reproducibles. La automatización de estos ensayos ha acelerado drásticamente la investigación y el desarrollo en campos como el descubrimiento de fármacos y la genómica funcional.
La Electroforesis en Gel: Una Herramienta Fundamental para el Análisis de Proteínas
Una vez que la concentración de proteínas ha sido determinada, una de las técnicas más comunes para analizar su pureza, tamaño y composición es la electroforesis en gel. La electroforesis es una técnica de separación ampliamente utilizada en biología molecular y bioquímica, que aplica una corriente eléctrica a moléculas biológicas cargadas, como proteínas, ADN o ARN, para separarlas en función de su tamaño, forma y carga eléctrica.
Principios de la Electroforesis
La electroforesis se lleva a cabo generalmente en una matriz porosa, como un gel de agarosa (para ADN grande) o poliacrilamida (para proteínas y ADN más pequeño), que actúa como un tamiz molecular. La muestra se carga en un extremo del gel, y se aplica un campo eléctrico. Las moléculas cargadas negativamente (como el ADN, que tiene un esqueleto de fosfato negativo, o las proteínas tratadas con SDS para adquirir una carga negativa uniforme) migran hacia el ánodo (electrodo positivo), mientras que las moléculas cargadas positivamente migran hacia el cátodo (electrodo negativo). La velocidad de migración a través del gel depende de varios factores:
- Tamaño: Las moléculas más pequeñas se mueven más rápido a través de los poros del gel que las moléculas más grandes, que encuentran más resistencia.
- Carga: Las moléculas con una carga neta más alta se moverán más rápido hacia el electrodo opuesto.
- Forma: Las moléculas compactas o globulares pueden moverse más fácilmente que las moléculas alargadas o desplegadas del mismo peso molecular.
Separación de Proteínas en Gel: SDS-PAGE
En el caso de las proteínas, la forma más común de electroforesis es la electroforesis en gel de poliacrilamida con dodecilsulfato de sodio (SDS-PAGE). Antes de la carga, las proteínas se tratan con SDS, un detergente aniónico que desnaturaliza las proteínas (las despliega) y las recubre con una carga negativa uniforme, enmascarando su carga intrínseca. Esto asegura que todas las proteínas migren hacia el ánodo y que su velocidad de migración sea inversamente proporcional a su tamaño molecular. Las proteínas más pequeñas migrarán más lejos en el gel, mientras que las más grandes quedarán retenidas en la parte superior.
La SDS-PAGE es fundamental para:
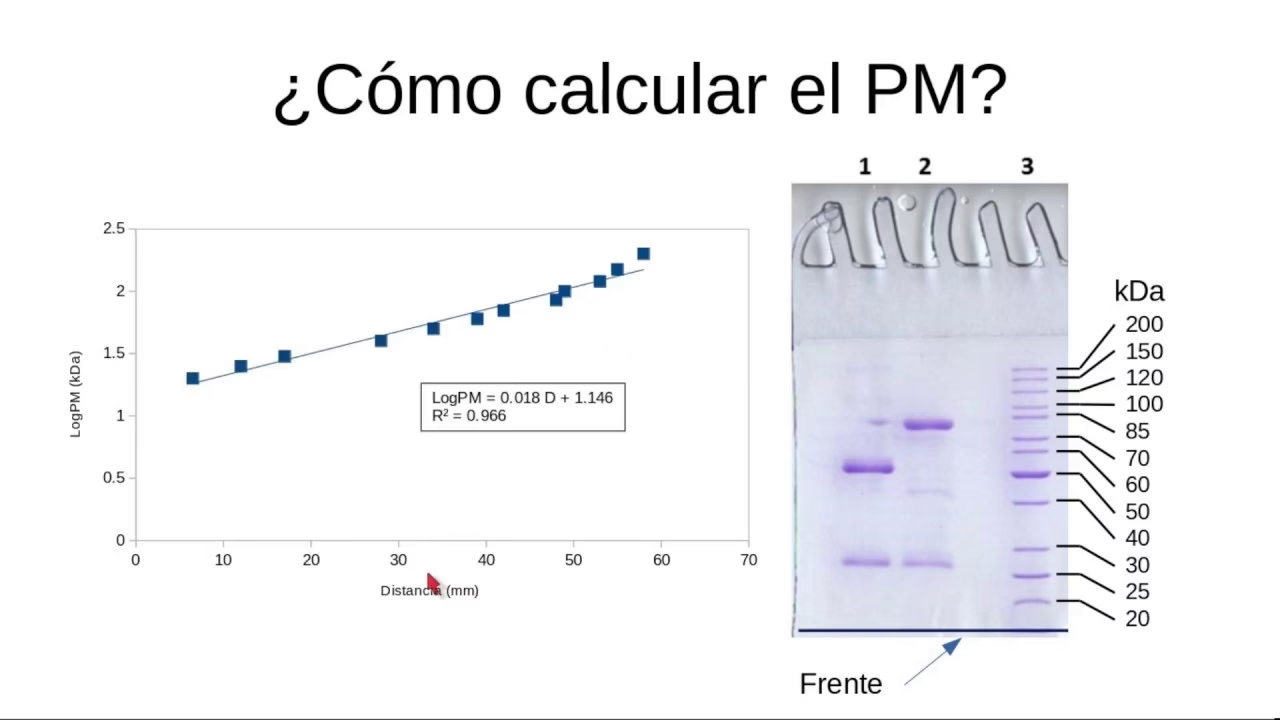
- Determinación del Peso Molecular: Comparando la migración de una proteína desconocida con un marcador de peso molecular (un cóctel de proteínas de tamaño conocido).
- Evaluación de la Pureza: Observando la presencia de múltiples bandas, que indicarían contaminantes.
- Análisis de Expresión: Comparando la abundancia de una proteína entre diferentes muestras.
- Detección de Modificaciones Post-Traduccionales: Que pueden alterar el peso molecular aparente o la carga de la proteína.
El Rol Crítico del TEMED y el APS en la Gelificación
La formación del gel de poliacrilamida, esencial para la electroforesis, depende de la polimerización de monómeros de acrilamida y bis-acrilamida. Este proceso es catalizado por un sistema iniciador que generalmente incluye persulfato de amonio (APS) y N,N,N',N'-tetrametiletilendiamina (TEMED).
La función del APS es generar radicales libres. Cuando el APS se disuelve en agua, el ion persulfato (S2O82-) se descompone lentamente para formar radicales libres de sulfato (SO4•-). Sin embargo, esta reacción es lenta por sí sola. Aquí es donde entra el TEMED.
El TEMED actúa como un catalizador de la polimerización al acelerar la descomposición del APS y, por lo tanto, la formación de radicales libres de persulfato. Es una amina terciaria que promueve la formación de estos radicales libres al reducir el persulfato. A su vez, estos radicales libres de persulfato, al añadirse a los monómeros de acrilamida, inician la formación de radicales libres de acrilamida. Estos radicales de acrilamida reaccionan entre sí para formar cadenas largas de poliacrilamida, que son entrecruzadas por la bis-acrilamida, creando la matriz tridimensional porosa del gel. La concentración de acrilamida y bis-acrilamida, junto con la cantidad de TEMED y APS, determina el tamaño de los poros del gel, lo que a su vez influye en la separación de las proteínas por tamaño.
Preguntas Frecuentes sobre la Cuantificación y Análisis de Proteínas
¿Cuál es el método más sensible para cuantificar proteínas?
Los métodos fluorimétricos, como los que utilizan colorantes específicos que se unen a proteínas (ej. Qubit), son generalmente los más sensibles, capaces de detectar proteínas en el rango de picogramos a nanogramos por microlitro. El ELISA también ofrece una altísima sensibilidad y, además, es específico para una proteína individual.
¿Se puede usar la electroforesis para cuantificar proteínas?
La electroforesis en gel (como SDS-PAGE) se utiliza principalmente para separar proteínas por tamaño y evaluar su pureza o presencia. Si bien se puede estimar la cantidad relativa de una proteína en una banda mediante densitometría (midiendo la intensidad de la banda con un software), no es un método de cuantificación absoluto para la concentración total de proteínas en una muestra. Para eso, se utilizan los métodos colorimétricos o fluorimétricos.
¿Qué es el TEMED y el APS y para qué sirven en el laboratorio?
El TEMED (N,N,N',N'-tetrametiletilendiamina) y el APS (persulfato de amonio) son reactivos clave utilizados para la polimerización del gel de poliacrilamida. El APS es el iniciador que genera radicales libres, y el TEMED actúa como catalizador para acelerar esta formación de radicales, que a su vez inician la polimerización de la acrilamida y bis-acrilamida para formar la matriz del gel utilizada en la electroforesis.
¿Cómo se determina el tamaño de una proteína en un gel de electroforesis?
El tamaño aproximado de una proteína en un gel de electroforesis (SDS-PAGE) se determina comparando la distancia de migración de la banda de la proteína de interés con la de un marcador de peso molecular. Este marcador es una mezcla de proteínas de tamaños conocidos que se carga en un carril adyacente. Al graficar la distancia de migración de las proteínas del marcador frente al logaritmo de sus pesos moleculares, se puede generar una curva estándar para estimar el tamaño de la proteína desconocida.
¿Qué factores influyen en la migración de las proteínas en un gel de SDS-PAGE?
En SDS-PAGE, donde las proteínas están desnaturalizadas y recubiertas con SDS para una carga negativa uniforme, el factor principal que influye en la migración es el tamaño molecular (peso molecular). Las proteínas más pequeñas migran más rápido y más lejos en el gel. Otros factores incluyen la concentración del gel (geles de mayor porcentaje separan mejor proteínas pequeñas, geles de menor porcentaje separan mejor proteínas grandes) y la fuerza iónica del tampón de corrida, aunque estos están estandarizados en la mayoría de los protocolos.
En resumen, la capacidad de cuantificar y analizar proteínas es indispensable en la investigación científica y el diagnóstico. Desde los métodos directos y rápidos hasta las técnicas colorimétricas y fluorimétricas de alta sensibilidad, cada enfoque ofrece ventajas únicas para determinar la concentración de proteína total. Complementariamente, técnicas como el ELISA permiten la cuantificación de proteínas específicas, mientras que la electroforesis en gel proporciona una herramienta poderosa para la separación y caracterización de proteínas por tamaño. La comprensión y la aplicación correcta de estas metodologías son esenciales para avanzar en nuestro conocimiento de los sistemas biológicos y desarrollar nuevas soluciones en medicina y biotecnología.
Si quieres conocer otros artículos parecidos a Desentrañando la Concentración de Proteínas: Guía Completa puedes visitar la categoría Cálculos.